PhD Dissertation: Multi-Omics in Research: Epidemiology, Methodology, and Advanced Data Analysis (2023)
Summary
Metabolomics, proteomics, and genomics analyses provide profound insight into human biology
and disease pathophysiology In this thesis, we explored the methodological challenges facing
these OMICs technologies and illustrated their applications in epidemiological studies. In part
one, we focused on some of the methodological challenges facing OMICs research. Chapter 2
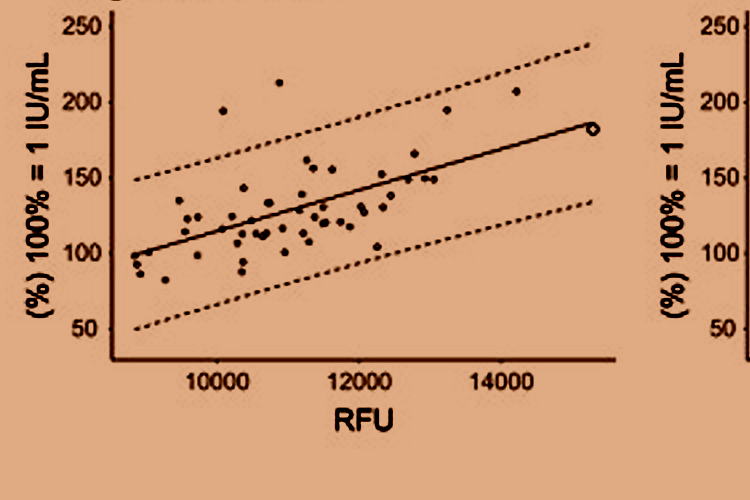
examined the agreement between aptamer-based proteomics measurements of venous thrombosis
(VTE) biomarkers with those measured by standardized clinical instruments. We reported
that 9 venous thrombosis biomarkers showed poor agreement with the clinical measurements.
The agreement was particularly poor for D-dimer, a marker often used in VTE diagnosis. In
Chapter 3, we explored the challenges of handling missing values in metabolomics data, particularly
from untargeted metabolomics platforms. We assessed the performance of two previously
reported methods for imputation, namely “k-nearest neighbor” (kNN) and “multiple imputation
using chained equations”, by simulating missing patterns in data from the NEO study. Our
findings showed that the multiple imputation method had less bias than the kNN method in
most scenarios, except for the scenario with small sample size and high level of missingness.
We further provided a publicly available R script to streamline the process of imputing missing
metabolomics data using the two methods.
Metabolites can provide unique insight into biological pathophysiology as they encompass the
cumulative effects of genetic, lifestyle, and environmental factors. Thus, metabolomics analyses
have been used to study biological processes such as aging. Some studies have used metabolomics
to estimate “metabolomic age”, a reflection of aging on a metabolomic level, using
prediction modelling methods. In Chapter 4, we addressed common challenges for developing
metabolomics-based age prediction models and developed a model to predict metabolomic
age using the Metabolon metabolomics platform measurements. We developed a model in the
INTERVAL study using ridge regression and bootstrapping. This study population is a relatively
healthy large population (n = 11,977) with a wide age range. Overall, after development and
internal validation, our prediction models for metabolomic age demonstrated good performance
(adjusted R2 = 0.83).
The second part of the thesis addressed various epidemiological research questions by utilizing
genomic data and metabolomics measurements (Metabolon and Nightingale platforms) and
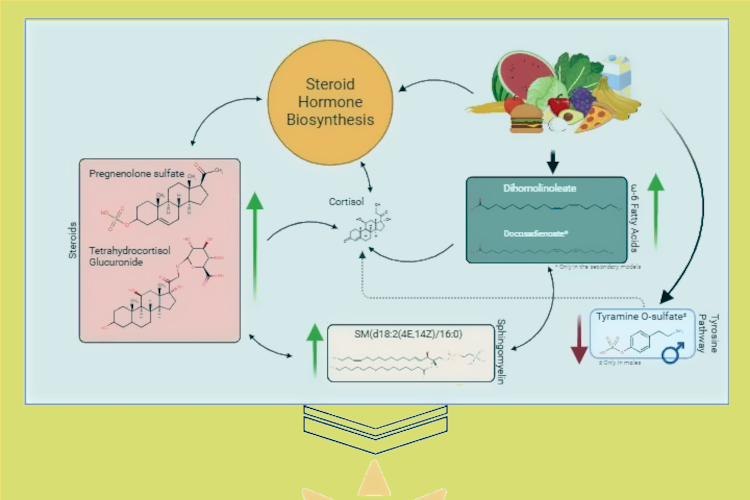
using advanced data analysis methods. In Chapter 5, we examined the associations of over 1300
metabolites, measured by the Metabolon platform, with hepatic triglycerides content (HTGC).
These associations included a range of endogenous metabolites from various pathways (particularly
amino acids and lipids), as well as novel and uncharacterized metabolites. Subsequently,
we constructed two complementary holistic networks, one biologically based and the other
statistically driven, to assign the associated metabolites to pathways. These interactive networks
revealed potential novel pathways associated with HTGC and possibly fatty liver diseases. We
further provided these networks on an online, publicly available webpage for further exploration
by the research community.
One type of variation in the genome is variation in the number of repeated cytosine-adenine-
guanine nucleotide sequences (CAG repeats). When the size of these repeats is beyond
specific pathogenic thresholds in or near specific genes, they lead to the onset of several severe
neurodegenerative diseases. One such case is the large CAG repeat sizes in the huntingtin (HTT)
gene leading to the onset of Huntington’s disease. Interestingly, recent studies have found that
CAG repeat sizes even within the non-pathogenic range are associated with health outcomes
such as cognitive function, depression, and body weight. In Chapter 6, we explored the possible
association between CAG repeat variation sizes below the pathogenic threshold (<36) in the
HTT gene and the metabolite levels measured by the Nightingale platform. Multilevel mixed-effects
and mediation analyses were conducted in pooled data from three large European cohorts
(n = 10,275). Our results showed that large non-pathogenic CAG repeat sizes were associated
with an unfavorable metabolomic profile, similar to that for an increased risk of cardiometabolic
diseases.
One of the advantages of metabolomics is the capability to measure “external” metabolites,
often referred to as xenobiotics, from medication use, diet, and environmental exposures and
contaminations. One such group of measured xenobiotics are the man-made per- and polyfluoroalkyl
substances (PFAS)—commonly known as “forever chemicals” due to their persistence in
the environment and the body. In addition to their persistent nature, high concentrations of PFAS
have been reported to be associated with severe health impact. Despite efforts to regulate their
production, PFAS levels remain detectable in the soil, drinking water, and human blood samples
in the Netherlands and Germany. In Chapter 7, we studied the associations between common
PFAS levels in blood, as measured by the Metabolon platform, and metabolomics and lipoprotein
profiles (Nightingale platform) of the general population in the Netherlands (NEO study) and
Germany (Rhineland study). Overall, PFAS metabolites were detectable in nearly all participants
of both studies. Furthermore, as confirmed by meta-analysis, PFAS levels were associated with an
unhealthy metabolomic profile characterized by increased fatty acids, low density lipoproteins,
and apolipoprotein B levels. These associations are reminiscent of the metabolomic profile for
increased risk of cardiometabolic diseases and were found to be particularly stronger in younger
individuals. These data indicate that PFAS are a health risk even in individuals that apparently
have not been exposed to high levels of PFAS.